Los resultados de recientes investigaciones, han expuesto fallas evidentes en la seguridad de los dispositivos médicos y en las regulaciones gubernamentales destinadas a proteger a los pacientes del mundo.
Las cifras son contundentes, más de un millón de pacientes se enfermaron o lesionaron y ochenta y tres mil fallecieron por los mismos dispositivos médicos que se suponía debían mejorar su salud. Información obtenida según informes conjuntos de medios de comunicación como NBC News y Associated Press, liderados por el Consorcio Internacional de Periodistas de Investigación ( ICIJ).
Estos informes, llamados los “Expedientes de Implantes”, representan el primer examen de la industria de dispositivos médicos y de sus guardianes en todo el mundo.

Entre los puntos claves del mismo, cabe destacar que existen hallazgos sobre un asunto más que crítico, y es que las autoridades sanitarias de todo el mundo presuntamente no protegieron a los pacientes de los dispositivos mal probados, incluso cuando la industria de dispositivos médicos obtuvo ganancias.
En respuesta a los avances del informe, la FDA se comprometió a proporcionar una red de seguridad de dispositivos médicos más robusta para los pacientes. El regulador aseguró que uno de sus enfoques específicos serán los dispositivos implantes de malla transvaginal, dispositivos vinculados a múltiples muertes, según informó MedTruth.
Reportes del ICIJ encontrados:
Los médicos, las autoridades sanitarias y los fabricantes de dispositivos retuvieron información médica vital de los pacientes. Unos 2,000 pacientes dijeron a ICIJ que sus médicos no les informaron sobre los riesgos para la salud de su dispositivo médico antes de que se implantaran.
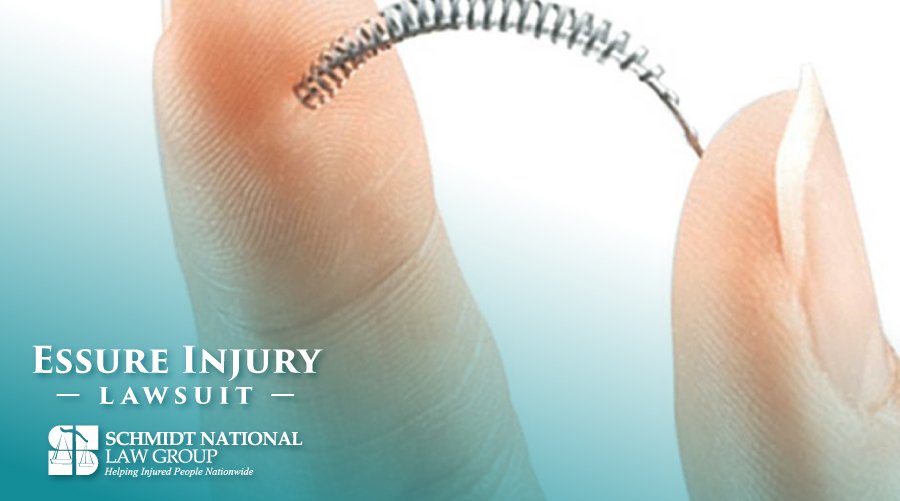
El anticonceptivo Essure está vinculado a miles de lesiones dolorosas, incluidas las perforaciones uterinas. Un análisis de ICIJ de los datos de eventos adversos en los EE. UU. Encontró casi 8.500 eventos en la última década que requirieron que el dispositivo Essure fuera retirara de los pacientes.
Los fabricantes de implantes mamarios enterraron evidencia de lesiones durante años, aprovechando la falta de eficacia de las normas de informes de la FDA. Antes de que la FDA instituyera normas de presentación de informes más rigurosas, el número promedio de sospechas de lesiones con implantes mamarios era menos de 200 por año hasta 2016. Con las normas más rigurosas, la cantidad de informes de lesiones aumentó a 4,567 eventos en 2017, y al menos 8,242 en los primeros seis meses de 2018.
¿Cómo se aprueban los dispositivos médicos?
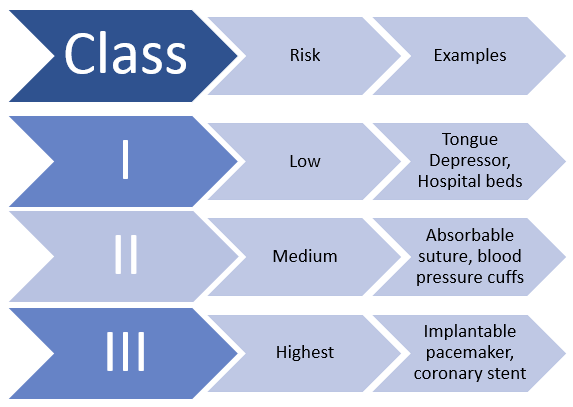
Los dispositivos médicos solo deben demostrar una “seguridad razonable” en los Estados Unidos antes de ser vendidos a los pacientes. Esta nivel bajo de evaluación, según el ICIJ, generalmente significa que los dispositivos se someten a un solo estudio y no a ensayos controlados aleatorios, una forma rigurosa de prueba que compara cómo se comportan los pacientes mientras se someten a diferentes tratamientos. En contraste, los fabricantes de medicamentos deben mostrar “pruebas sustanciales” de la seguridad y efectividad de un nuevo producto, lo que generalmente requiere tres ensayos controlados aleatorios. Como informó el ICIJ, “En 2017, la FDA aprobó más de tres veces más dispositivos que en 2010, mientras que sus advertencias a los fabricantes de dispositivos sobre la seguridad del producto se redujeron en casi un 80 por ciento”. La FDA reconoce abiertamente que aprueba una docena de dispositivos médicos nuevos o modificados todos los días hábiles.
¿Cómo prevenir estos efectos?
Es muy importante reportar, reconocer y analizar la información acerca de los eventos adversos relacionados con todos los dispositivos médicos del mercado. La FDA suministra en este portal la subscripción sin costo vía email de diferentes alertas relacionadas con DM.
Más allá de lo anterior y debido a que pese a los filtros regulatorios será muy posible encontrar tecnologías que fallarán durante su proceso de uso, gran parte de la gestión de seguridad debe estar a cargo del personal relacionado con procesos asistenciales, el personal de ingeniería clínica y hasta los mismos usuarios junto con sus familias.
Es nuestro deber gestionar, consultar y obtener la mayor cantidad de información sobre cualquier dispositivo implantable:
- Quién es el Fabricante?
- Qué tipo de dispositivo es y qué riesgos asociados o advertencias existen?
- Eventos adversos asociados al DM.
- Procedimientos de calidad y controles realizados.
- Registro sanitario.
- Procesos de seguimiento al paciente y soporte técnico del fabricante.
- Información clara del médico responsable del procedimiento asociado al dispositivos.
- Entre otros.
Para el ambiente hospitalario y compañías comercializadoras existen también herramientas de software que pertimen realizar la gestión y registrar toda la trazabilidad de sus dispositivos médicos.
Si estás interesado en conocer de que se trata has click aquí: http://latam.qsystems.com.co/keeper